
近日,复旦大学智能复杂体系基础理论与关键技术实验室孙思琦团队与香港中文大学、耶鲁大学联合团队提出了一种全新的超高速、高灵敏度的蛋白质同源物检测方法——Dense Homolog Retriever (DHR)。该方法利用蛋白质语言模型和密集检索技术,在不依赖序列比对的情况下,实现了蛋白质远程同源物的快速检测,显著提升了多序列比对(MSA)构建速度和蛋白质结构预测效率。
目前,该成果相关论文Fast, sensitive detection of protein homologs using deep dense retrieval已发表于生物技术领域顶级期刊Nature Biotechnology。

蛋白质同源物检测的基础与挑战
蛋白质同源物检测是计算生物学中的一项基础工作,在蛋白质结构预测、生物分子功能分析、转录调控研究、系统发育重建以及生物标志物预测和药物发现等几乎所有生物序列相关研究中都发挥着重要作用。随着下一代测序技术的发展,生物序列数据库的规模不断扩大,传统的同源物检测方法在速度和灵敏度之间难以权衡,常常会遗漏远同源蛋白(即序列相似性较低但结构或功能相似的蛋白质)。这些方法中的一部分会在第一阶段就过于激进地丢弃远同源序列,剩下的方法则非常依赖于序列比对,耗时费力。
通过语言模型实现快速的蛋白质同源物检测
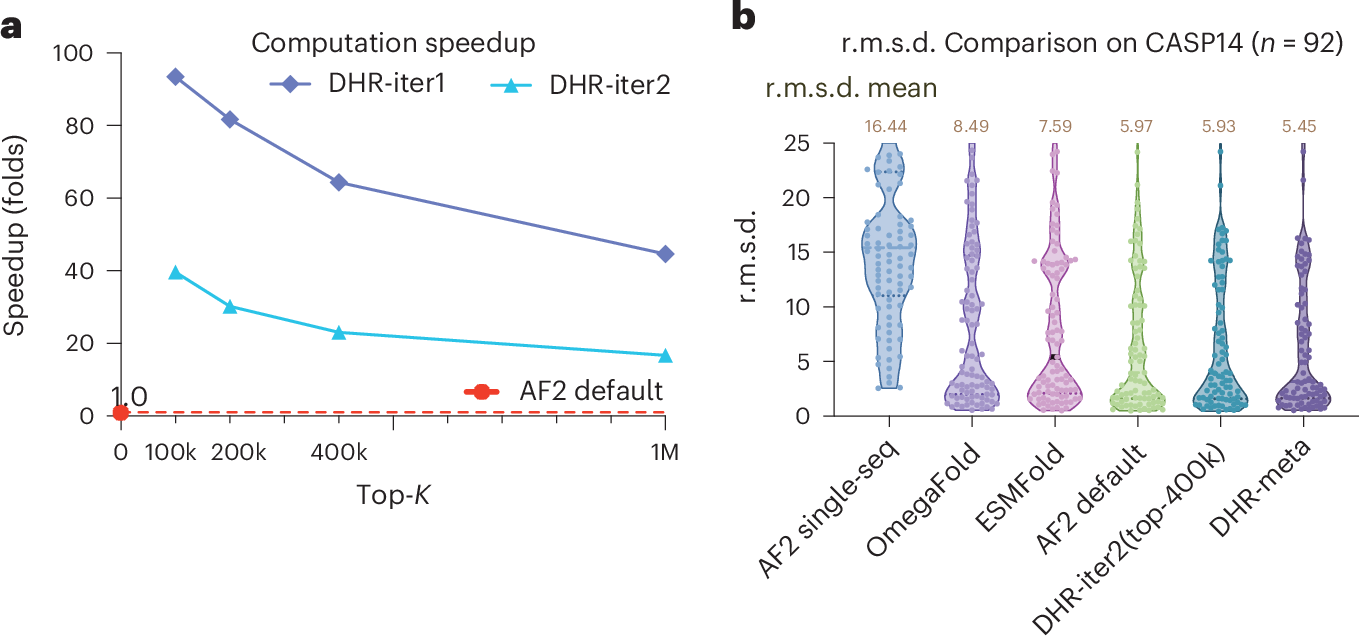
图1DHR用于快速检测远程同源物。a)与AF2使用的传统MSA构造方法相比,DHR实现了极快的同源物搜索和MSA构建。b) DHR构建的MSA能提升结构预测精度。
似于基于人类语言训练的ChatGPT,在大规模序列数据集上预训练的蛋白质语言模型展现出捕获进化信息的潜力。此外,蛋白质语言模型的另一个优势在于其无需比对的特性,在处理序列时速度极快。因此,联合团队基于蛋白质语言模型和密集检索(Dense Retrieval)开发了Dense Homolog Retriever (DHR)方法(图1)。该系统利用先进的蛋白质语言模型将查询序列编码为嵌入式向量表示,并通过向量之间的简单相似度度量对数据库进行搜索和同源性比较。
实验表明,DHR在远程同源物检测中表现出极高的速度和灵敏度。与传统方法相比,DHR的灵敏度提高了10%以上,在传统方法难以识别的超家族水平上,DHR的灵敏度更是提高了超过56%。同时,DHR的速度是传统方法(如PSI-BLAST和DIAMOND2)的22倍,是HMMER的28,700倍。当将DHR与JackHMMER串联以加速迭代MSA构建过程时,DHR比传统方法快93倍,并且构建的MSA与AlphaFold2生成的MSA高度一致。此外,DHR与JackHMMER生成了更多样化和全面的MSA,在与AlphaFold2生成的MSA合并时,平均能提高0.4 Å的蛋白质预测精度。
总结和展望
DHR为蛋白质远程同源物鉴定这一项计算生物学的基本挑战提供了强大的解决方案,有望成为蛋白质进化、结构和功能分析的基础,这也是语言模型在生物学中的一种强大应用。除了预测蛋白质结构和功能外,还可以进一步开发此类模型以解决序列分析中的其他重要计算挑战。联合团队后续将利用这些方法无需序列比对的特性和处理海量数据集的能力,开发更强大的工具。
论文链接:https://www.nature.com/articles/s41587-024-02353-6
此外,近日孙思琦团队还与香港中文大学、麻省理工学院、智峪生科联合团队提出一种创新的高精度端到端RNA三维结构预测方法——RhoFold+。该方法基于RNA语言模型和多序列比对数据,通过充分利用RNA海量序列数据资源,实现了RNA三维结构的高效预测。
目前,该成果相关论文Accurate RNA 3D structure prediction using a language model-based deep learning approach已发表在方法学领域顶级期刊Nature Methods。

RNA结构预测的挑战与机遇
RNA分子在分子生物学中扮演重要角色,在基因表达、蛋白质合成、细胞代谢和信号传导等生命活动中承担着多种关键功能。RNA结构对于理解其生物功能及相关药物开发至关重要,但由于其结构灵活性,实验测定三维结构具有挑战性。目前研究人员开发的计算方法各有局限:基于多序列比对(MSA)的方法(如AlphaFold3)准确度高但耗时较长,而基于单序列的方法(如DRFold)虽快速但准确性较低。
通过语言模型实现精确的RNA结构预测
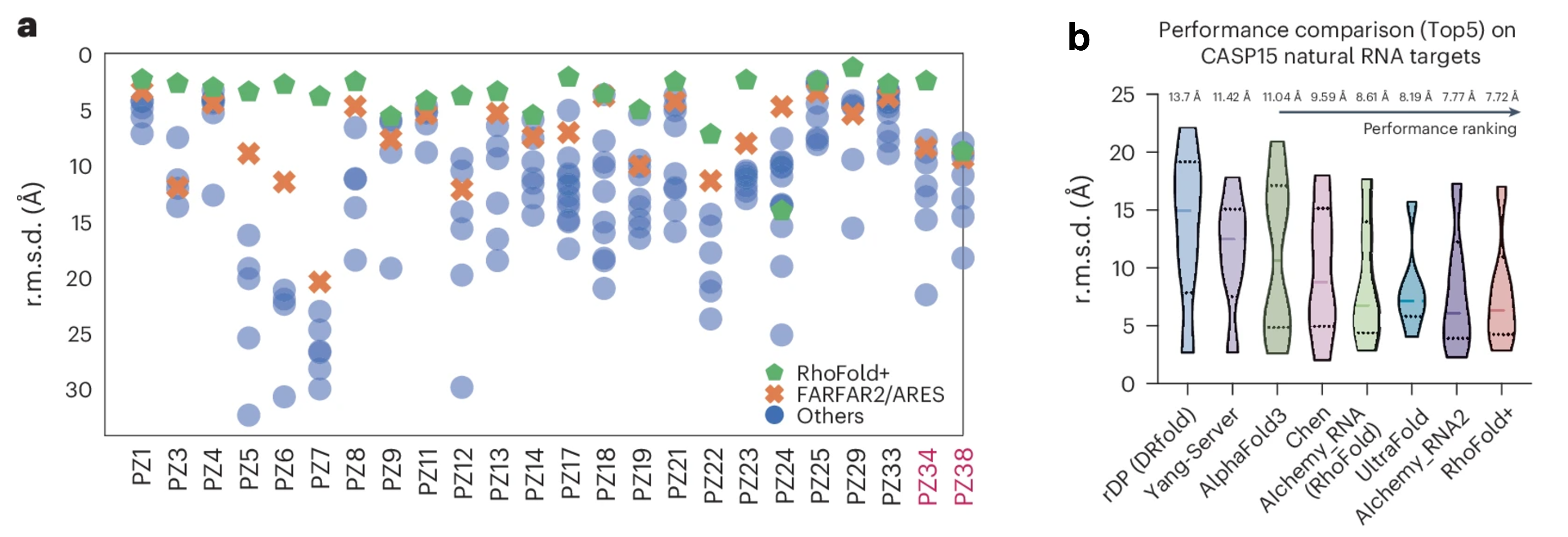
图1RhoFold+实现高精度RNA结构预测。a)在RNAP-Puzzles基准测试集上,RhoFold+的平均RMSD达到4Å,显著优于现有最佳方法。b)在CASP15测试集的评估中,RhoFold+的预测精度超越了包括AlphaFold3和基于专家知识的AIchemy_RNA2在内的其他方法。
RhoFold+通过整合近2400万条RNA序列数据预训练的语言模型RNA-FM,与多个深度学习模块相结合,能够从RNA序列直接预测其三维结构。RhoFold+的架构包含两个核心模块:特征处理模块和结构预测模块。特征处理模块利用RNA-FM生成的序列嵌入信息,并结合多序列比对(MSA)数据,提取序列中的进化保守信息。结构预测模块则通过几何感知注意力机制和生物学约束,在三维空间中精确预测RNA的全局构象。
在国际RNA结构预测竞赛RNA-Puzzles中,RhoFold+展现出优异性能,其预测结构与实验结构的平均RMSD(均方根偏差)为4.02Å。除PZ24外,RhoFold+在所有测试目标上的表现均优于其他竞争方法(图1)。此外,RhoFold+具有显著的计算效率,能在约0.14秒内完成典型RNA-Puzzles目标的结构预测。在另一个权威竞赛CASP15中,RhoFold+的预测精度比第一名AIchemy_RNA高0.06Å,尽管后者依赖专家知识辅助。RhoFold+还显著超越了其前代模型RhoFold(AI组第一名),预测精度提升了1Å。
总结与展望
RhoFold+的成功开发不仅提高了RNA三维结构的预测准确性,更通过其高效的处理速度,为大规模RNA结构分析提供了可能,这对药物开发、合成生物学和基因调控等领域具有重大意义。未来,研究团队还将进一步开发此类模型,以支持对复杂RNA,孤儿RNA和RNA复合物等结构预测。通过这些改进,RhoFold+将为RNA生物学领域带来更强大的计算工具,加速从RNA结构到功能的机制研究。
论文链接:https://www.nature.com/articles/s41592-024-02487-0









